(IDWR 2002年第24号掲載)
クロイツフェルト・ヤコブ病(Creutzfeldt‐Jakob disease, CJD)は100万人に一人の割合で弧発性または家族性に生じ、脳組織の海綿(スポンジ)状変性を特徴とする疾患である。CJD は1920 年代初頭、ドイツの神経病理学者Creutzfeldt とJakob によって記述された。現在では成因から、プリオン(prion)病、また病理から伝達性海綿状脳症(transmissible spongiform encephaolopathy, TSE)として哺乳類の神経疾患群にひとくくりにされている。近年、プリオン病またはTSE の感染性がクローズアップされ、社会的に認知された。
プリオン
Prion とは蛋白質性感染粒子(proteinaceous infectious particle)のことで、TSE の核酸を含まない感染性病原体をさす造語で、米国のPrusiner 博士(1997 年のノーベル賞受賞者)によって1982 年に提唱された。Prusiner 博士は10 年の歳月をかけ、プリオン病の罹患脳から幅4nm 、長さ数百nm程度の感染性の微細線維状物質(エイズの原因ウイルスHIV‐1 は直径100nm で球状)を濃縮していき、プリオン説を唱えるに至った。この微細線維状物質は現在、宿主プリオン蛋白が異常構造体へ変換され、凝集することによって形成さ れていると考えられている。
ヒトプリオン病
プリオン病では、異常構造を有する異常プリオン蛋白が中枢神経系に蓄積し、不可逆的な致死性神経障害を生ずる。ヒトプリオン病の大半を占めるのは弧発性 CJDである。プリオンには感染性があり、感染性ヒトプリオン病としてクールー(Kuru)、(新)変異型CJD ((new)variant CJD, vCJD)、移植後CJD がある。クールーはニューギニアの高地に住むFore 族に年間1%の高率で発症していた疾患である。1966 年にGadjusek らがチンパンジーへの感染実験に成功した。彼らは続いてCJD の感染実験にも成功し、Gadjusek は1976 年にノーベル賞を受賞した。
1996 年に英国で発表され、ヨーロッパ及び世界中をパニックに陥れたのはvCJD である。これは牛海綿状脳症(bovine spongiform encephalopathy, BSE )に起因していると考えられている。
一方、プリオン遺伝子に変異を持ち、異常プリオン蓄積の原因となる疾患に遺伝性CJD 、クールー斑状沈着を特徴とするゲルストマン・ストロイスラー・シャインカー症候群(Gerstmann-Str ?ussler-
Scheinker syndrome, GSS)、また致死性家族性不眠症(fatal familial insomnia, FFI)などがある。
動物のプリオン病
動物のプリオン病には、18 世紀にはすでに知られていた羊のスクレイピー(scrapie)、シカ慢性消耗病(chronic wasting disease,CWD )、ミンク伝達性脳症(TME)、1987 年に発表され日本でも4頭見つかっているBSE 、またネコ海綿状脳症(FSE)などがある。ネコや動物園のチータなどのTSEは、BSE 由来の餌が原因であると考えられている。
プリオン蛋白の伝達性獲得機構
プリオン病の病因は、神経細胞表面にある正常プリオン蛋白が異常構造体へ変換後、異常プリオン蛋白の蓄積が生じ、神経細胞が変性した結果であると言われ ている。プリオン蛋白(prion protein, PrP)はさらに正常型(細胞型;cell)プリオン蛋白(PrPC)と、scrapie のSc を使用して異常(感染)型プリオン蛋白(PrP Sc)に分類されている。PrPC はC 末端部で細胞膜へ連なり、PrPC からPrP Sc への変換は細胞膜上、または細胞質への再取り込み後におこると信じられている。PrP Sc の形成は蛋白のPrPC の構造的な変換によって生じるので、PrP Sc の集積にはPrP C の存在が不可欠である。PrP C に多いらせん状構造(αへリックス)が板状構造(βシート)へ変換した結果、PrP Sc になる。この構造変換によってプリオン蛋白は伝達性を獲得する。
異常構造の伝達は種々の宿主因子が関与しながら、異常構造体を核として正常プリオン蛋白が変換され凝集体が形成されていく数種のモデルによって説明され ている。この構造変換に伴い、プリオン蛋白は伝達性に加え蛋白分解酵素耐性を獲得する。蛋白分解酵素耐性獲得のメカニズムを理解するには蛋白分解酵素をは さみとして考えると解りやすい。はさみによる切断には、対象が板状(βシート)であるよりもらせん状(αへリックス、ひも状)である方がむいており、プリ オン蛋白の構造変化が蛋白分解酵素耐性を生じていると想像できる。
疫 学
我が国を含め、世界各国の弧発性CJD 有病率は同一で、人口100 万人対1 前後とまれな疾患である。この様に、地理的に違いがない感染症としてもCJD は特異的である。発症年齢の平均は62 歳であり、女性が男性よりやや多い。大多数が弧発例で、家族性あるいは遺伝性のGSS が約10% ある。
vCJD は2002 年5 月までに英国で122 名報告されており、今後の推移予測には数千から数万台 の幅がある。他にフランスで6 例、アイルランド、イタリア、米国、香港でそれぞれ1 例報告されてい る。vCJD のリスクをふまえ、わが国では2001 年3月より英国、アイルランド、スイス、スペイン、ドイツ、フランス、ポルトガル、ベルギー、オランダ、イタリアに1980 年以降、通算6カ月以上の滞在歴を有する人の献血は受け付けないことになっている。
我が国の感染症発生動向調査によるCJD の報告は、1999 年4 〜12 月に87 例、2000 年1 〜12 月に102 例、2001 年1 〜12 月に130 例(暫定データ)となっている。
臨床症状
弧発性CJD の主症状は進行性痴呆とミオクローヌスである。発病より数ヶ月で痴呆、妄想、失行が急速に進行し、筋硬直、深部腱反射亢進、病的反射陽性などが認められ る。さらに起立歩行が不能になり、3 〜7 カ月で無動性無言状態に陥る。1〜2年で全身衰弱、呼吸麻痺、肺炎などで死亡する。
遺伝性CJD は弧発性CJD に似た臨床症状を示す。GSSは小脳性失調とその後の痴呆を特徴とする。
vCJD は20歳代の若年者に好発し、行動異常、感覚障害、ミオクローヌスを主症状とし、無動性無言状態に陥るのに1年を要する。
病原体
プリオンは主に、異常プリオン蛋白の凝集による幅4nm 、長さ数百nm 程度の感染性微細線維状物質からなり、その感染価は得られた臓器により一致しないことがあることから、プリオン以外に感染性に影響する因子が想定されている。
CJD では一般に空気感染や経口感染はないとされている。vCJD 、BSE では病原体の経口摂取による感染が疑われている。紫外線、エタノールなどの消毒法が無効であり、手の汚染、注射針などの刺傷、感染物の眼への飛沫や手で眼 をこすることなどをさける。汚染したものは焼却するか、SDS (sodium dodecyl sulfate)を3%含む溶液中で100度、5分間以上加熱処理する(消毒法の詳細については後述)。
臨床材料はバイオセーフテイレベル2 (BSL‐2)において扱う。プリオン病原体などの臨床材料または剖検材料からの抽出は、BSL‐ 2 内の安全キャビネット内で行う。
病原診断
異常プリオン蛋白は上記のように蛋白分解酵素に耐性を獲得するので、剖検材料(脳組織、扁桃、脾、髄膜、移植例では角膜)から蛋白分解酵素耐性の異常プ リオン蛋白の同定をウエスタンブロット法やELISA 法によって行う。また、蟻酸処理後に抗プリオン抗体による免疫染色を行う。わが国の食肉衛生検査所では、食肉処理を行う全てのウシの延髄乳剤をサンプルと して、ELISA 法によってスクリーニングを行 い、病理組織および免疫組織化学検査とウエスタン ブロット法によってBSE の確定検査が行われている(2001 年10 月より実施)。実際に感染性を調べる高感度なバイオアッセイとして、正常プリオン蛋白を過剰に発現させたトランスジェニックマウスが開発され、短期間で発 症するので有用である。脾臓には感染後40 日程度で異常プリオン蛋白の集積が認められるので、有効な検索対象であることが判明している。東北大の北本哲之教授によって開発されたマウスプリオン遺伝 子をヒト型へ変更したノックインマウスも、今後活用されていくであろう。他に、尿、血液を使用した検査系の開発も進められている。
 |
 |
|
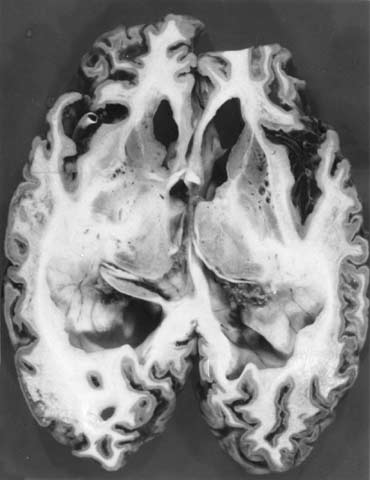
写真1. CJD 解剖例の脳割面(北大分子細胞病理、長嶋和郎教授供与) |
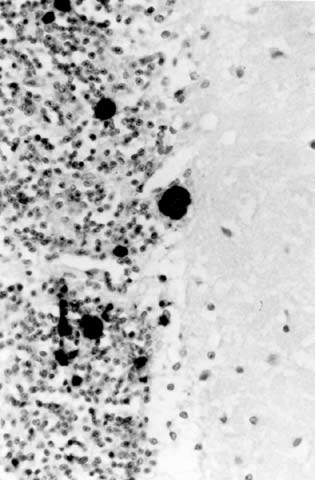
写真2. 異常プリオン蛋白からなるアミロイド斑を免疫染色した例(北大分子細胞病理、長嶋和郎教授供与) |
脳波は初期から基礎律動の不規則性がみられ、その後高振幅鋭徐波(PSD )が出現するのがCJD の特徴である。脳CT 画像上では初期に軽度の大脳皮質の萎縮、脳室拡大がみられ、その後急速な大脳、 小脳の萎縮、著明な脳室拡大、白質のびまん性低吸収域が認められる。CJD 解剖例(北大分子細胞病 理、長嶋和郎教授供与)の脳割面(写真1)では、大脳、小脳の脳溝拡大と全脳室の拡大が高度であり、 大脳、小脳皮質が薄くなる。vCJD 例では脳波のPSDはみられず、MRI で視床枕の高電子密度が特異的所見であると報告された。
解剖脳の病理検索では皮質の萎縮、特有の海綿状変化、神経細胞の脱落、アミロイド斑など が指標となる。異常プリオン蛋白からなるアミロイド斑を抗プリオンペプチド抗体によって免疫染 色した例(北大分子細胞病理、長嶋和郎教授供与)を示す(写真2)。 小脳顆粒層の境界部に抗 体によって染色された部分を認める。PCR によるゲノムの解析では、血液などから抽出したゲノ ムDNA をもとにプリオン遺伝子のシークエンスを決定する。日本人の遺伝性プリオン病では、東 北大の北本哲之教授らによってコドン102 、105 、145 、178 、180 、198 、200 、210 、217 、232 などに変異が発見されている。
ホルマリン固定後の蟻酸不活化処理パラフィン包埋組織については危険性がなく、室温における輸送が可能である。3%SDS 中で5 分間以上煮沸したウエスタンブロット法のサンプルに感染性はない。器具などの汚染の不活化・消毒は困難である。消毒法としては、焼却あるいは3%SDS中 で5 分間煮沸、5%次亜塩素酸ナトリウム中に2 時間以上、あるいは2N NaOH に1 時間、室温で浸す。高圧蒸気滅菌(オートクレーブ)は132 ℃で1 時間行うが、乾燥した器具などには適さない。
治療・予防
治療法は現在開発されておらず、対症療法が主体である。栄養の補給、関節拘縮、褥瘡、気道、尿路感染などに注意する。最近、クロルプロマジンやキナクリ ンなどの投与が行われ、一時的に症状の改善が得られたとする報告があるが、治癒するものではなく、今後の研究成果に期待がかかっている。
ヒツジの脳はフランスで数百年に渡り食されており、スクレイピーのヒトへの伝達は起こらないと推定されている。しかし、ヨーロッパにおいてヒツジ及びヤ ギにBSE が伝達している可能性が否定できないため、ヨーロッパにおけるヒツジ及びヤギ神経組織の摂食にも注意が必要である。
感染症法における取り扱い (2012年7月更新)
全数報告対象(5類感染症)であり、診断した医師は7日以内に最寄りの保健所に届け出なければならない。
届出基準はこちら
【参考資料】
厚生省保健医療局疾病対策課編クロイツフェルト・ヤコブ病診療マニュアル 1997 年
(国立感染症研究所感染病理部 高橋秀宗、佐多徹太郎)